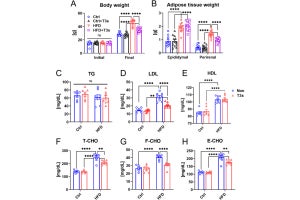
βUCP2Tgマウスは、高血糖においてインスリンの分泌が減り、ヒトの糖尿病に似た病態が示され、膵β細胞でUCP2が増えると、インスリン分泌に重要なATPが減少したが、脱共役は糖尿病がないマウスと同等であることが確認されたという。
また、UCP2が解糖系酵素「アルドラーゼB」を増やすことも見出されたとする。アルドラーゼBは主に肝臓に存在し、果糖の代謝に関わることが知られているが、膵β細胞での役割は不明であったという。今回の解析から、膵β細胞においてアルドラーゼBが増えると、UCP2が増えたときと同様に、ミトコンドリア機能異常、インスリン分泌減少が生じることが判明したとする。
さらに、膵β細胞からのインスリン分泌は、カルシウムにより調節されることが知られているが、UCP2やアルドラーゼBが増えると、カルシウムを貯蓄する細胞内小器官である小胞体から細胞質へのカルシウム供給が減ることが確認されたともするほか、これらのミトコンドリア機能低下やカルシウム供給異常は、アルドラーゼBを抑制することにより回復したとする。
-
今回の研究成果のイメージ (出所:群馬大Webサイト)

なお、研究チームではこれらのマウスにおいて認められたUCP2によるインスリン分泌の減少が、ヒト膵島においても観察されることを確認したとしており、今回の成果である糖尿病の膵β細胞で増えるUCP2が、アルドラーゼBを介してミトコンドリア機能低下および小胞体からのカルシウム供給異常を誘導し、膵β細胞からのインスリン分泌低下を引き起こすことで、血糖値のさらなる上昇を招くという新たな機構が解明されたことは、今後、UCP2もしくはアルドラーゼBを標的としたインスリン分泌を回復させる糖尿病治療への展開につながることが期待されるともしている。