熊本大学と千葉大学は5月16日、ヒトや出芽酵母などの6つのモデル生物の約40万件のデータを収集し、エピゲノム統合データベース「ChIP-Atlas」のメジャーアップデートを行い、ゲノムの三次元構造・疾患感受性ゲノム変異などの注釈づけ情報を統合したことに加え、遺伝子発現制御に関わるエピゲノム状態の変容を検出する比較解析ツールも実装したことを共同で発表した。
同成果は、熊本大 生命資源研究・支援センターの鄒兆南助教、同・沖真弥教授、千葉大 国際高等研究基幹/同・大学大学院 医学研究院の大田達郎准教授らの共同研究チームによるもの。詳細は、核酸に関する全般を扱う学術誌「Nucleic Acids Research」に掲載された。
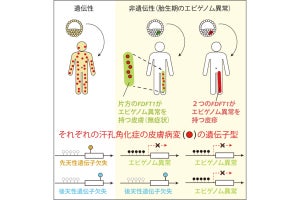
ヒトの身体中のすべての細胞が同じゲノムを持つのに、機能や役割の異なる細胞が何種類も存在するのは、細胞の種類ごとに特異な遺伝子セットを活性化する遺伝子発現制御機構が備わっているからだ。ゲノム上に存在するエンハンサ領域の活性化、DNAのメチル化状態、そしてゲノムに結合する約1000種類の転写因子が互いに協調することで、DNAの塩基配列を変えることなく、遺伝子の機能が決められている。この仕組みが「エピゲノム」と呼ばれる。
これまでに、ゲノム上の転写因子結合場所を調べる「ChIP-seq」、転写因子が結合しうる活性化エンハンサ領域を調べる「ATAC-seq」、DNAのメチル化状態を調べる「Bisulfite-seq」などのシーケンス技術が広く利用され、数十万件のエピゲノムに関する実験データが報告されてきた。しかし、それらの膨大なデータの意味を理解して活用するためには、高度な解析技術と豊富な計算資源が必要だという。そこで研究チームが開発したのが、国立遺伝学研究所のスーパーコンピュータを利用し、ヒト、マウス、ラット、ショウジョウバエ、線虫、出芽酵母の6つのモデル生物の約40万件の上述の3種類のシーケンス技術のビッグデータを収集・計算した成果であるChIP-Atlasだ。ChIP-Atlasは、世界最大規模のエピゲノム統合データベースという地位を確立したことから、研究チームは今回、機能拡充を含め、正式に論文として報告することにしたという。
-
ChIP-Atlasの公開データ(出所:共同プレスリリースPDF)

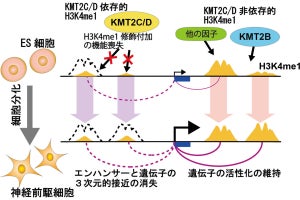
ChIP-Atlasによる活性化エンハンサ領域とDNAメチル化状態の可視化により、これまでは多大なコストが必要とされていたエンハンサの特定を、Web上の容易な操作で予測できるようになり、また推定されたエンハンサ領域に結合する転写因子も視覚的に理解できるようになった。しかし、これだけでは1つ1つのエンハンサがどの遺伝子の発現制御に関わるかを特定することができない。これは、ゲノムは三次元構造を形成しているため、エンハンサはその最も近傍の遺伝子を標的としているとは限らないからだ。それを可視化するため、今回、ChIP-Atlasに「Annotation Track」として、ゲノムDNAの三次元構造が調べられた実験データが統合された。
さらに今回のアップデートでは、疾患に関連するゲノム上の一塩基多型(SNP)などの変異情報も、Annotation Trackに新たに追加された。これにより、エンハンサ領域の位置情報に加え、そこに存在する疾患関連SNPの情報を加味することで、疾患の背景にある遺伝子発現制御機構を一括で理解できるようになったという。これにより、ゲノム変異に起因する疾患の発症メカニズムに関するリーズナブルな仮説を形成することが可能となったとする。
-
疾患関連SNP周辺エピゲノム状態の可視化(出所:共同プレスリリースPDF)

また、生体内イベントのメカニズムに迫るためには、その分子基盤であるエピゲノム状態の変容を正確に評価することが重要だが、これにもやはり情報学・統計学の知識と潤沢な計算資源が必要。そこでChIP-Atlasの新機能として、3種類のビッグデータの比較解析を支援するオンライン解析ツール「Diff Analysis」が実装された。
-
エピゲノム状態の皮革解析ツール(出所:共同プレスリリースPDF)

同ツールは、ChIP-Atlas上にある数十万件の解析済み実験データを扱っているため、Web上で2群の実験データのアクセス番号を入力するだけで、わずか数分で計算が終了し、比較結果を閲覧できるようになる。
今回のメジャーアップデートにより、疾患ゲノム情報をはじめとするゲノム・エピゲノム注釈づけデータが大幅に拡充され、ChIP-Atlasは遺伝子発現制御の異常に起因する遺伝性疾患の成り立ちを解明するデータ基盤へと進化したとしている。