東京大学生産技術研究所(東大生産研)、立教大学、みずほ情報総研(みずほ)、神戸大学、国立医薬品食品衛生研究所、NECの6者は4月30日、これまで主に理論創薬の分野で用いられてきた「フラグメント分子軌道(Fragment Molecular Orbital:FMO)法」をナノ-バイオ複合系に適用する技術を新たに開発し、シリカ表面と「ペプチド」(微小タンパク質)の相互作用の大規模モデリングに応用することに成功したと共同で発表した。
成果は、立教大理学部の望月祐志教授、みずほの福澤薫チーフコンサルタント、東大生産研の沖山佳生特任研究員、同・渡邉千鶴特任研究員らの共同研究チームによるもの。研究は東大生産研を拠点として行われている、文部科学省次世代IT基盤構築のための研究開発「イノベーション基盤シミュレーションソフトウェアの研究開発」プロジェクトよるもので、詳細な内容は、4月12日付けで「Chemical Physics Letters」に掲載された。
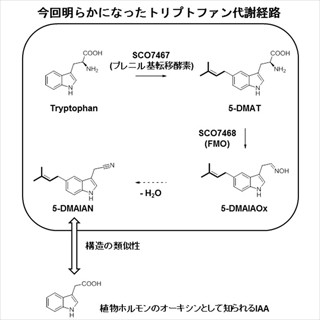
FMO法とは、現・神戸大大学院 システム情報学研究科の北浦和夫特命教授らが1999年に提案した、生体分子系に対する効率的な計算手法のことである。巨大系を比較的小さなフラグメント(アミノ酸残基など)に分割し、各フラグメントの「モノマー(フラグメント単体)」(画像1)と「ダイマー(フラグメントペア)」(画像2)の分子軌道計算を並列処理することにより、全系の電子状態をこれまでの手法よりはるかに短時間に高精度で解析することができる近似計算法だ。

|

|
|
画像1。モノマーのイメージ(プレス発表用資料から抜粋) |
画像2。ダイマーのイメージ(プレス発表用資料から抜粋) |
FMO法は大規模分子系を量子論的に扱うことが可能であり、計算からは、フラグメント間の「相互作用エネルギー(Inter Fragment Interaction Energy:IFIEもしくはPair Interaction Energy:PIE)」が得られるため、これまで生化学や生物物理化学、とりわけ創薬分野での応用計算が盛んに行われている。
これは、タンパク質のアミノ酸残基と薬剤分子との相互作用を、IFIEを指標にして詳細に解析することが直截に可能であり、薬剤分子とタンパク質の結合様態の理解、さらには薬剤の骨格・置換基の改変による最適化に好適であるためだ。
一方で、固体と生体分子の相互作用は、インプラントの表面改質、微小粒子による薬剤投与(ドラッグデリバリ)などの医療工学、あるいは生体結晶析出(バイオミネラリゼーション)などの医療工学、生物工学などいわゆるナノ-バイオの境界領域の問題として近年重要視されている。特に、特定組成の固体面を認識する人造ペプチドは世界中のグループが研究開発を進めており、日本ではがん研究所の芝清隆氏(蛋白創製研究部部長)らが先端を走っている。
こうした中、研究チームでは独自開発の「ABINIT-MPプログラム(MIZUHO/ABINIT-MP)」を拡張して4体までのフラグメント展開を行う「FMO4法」を実装し、4体補正(4体展開)によって計算精度が飛躍的に向上することを実証した。
一般的なFMO法が2体展開なのに対し、FMO4法はフラグメントの展開を「テトラマー(4体)」(画像3)まで拡張することにより、「リガンド」(特定の受容体に特異的に結合する物質)の複数分割とアミノ酸残基の主鎖・側鎖分割を伴う官能基単位の相互作用解析が可能となり、高い空間高解像度と定量製を両立させた次世代のIFIE解析を可能とするアプローチだ。一般的なFMO法とは異なり、FMO4計算ではダイヤモンドやシリコンなどの3次元の結合ネットワークを持つ固体をも扱うこともできることから、製薬企業からも注目を集めている。

|
|
画像3。テトラマーのイメージ(プレス発表用資料から抜粋) |
今年度は、ABINIT-MPの高速化(NECと共同)や解析手法の洗練(神戸大学の田中成典教授、国立医薬品食品衛生研究所の中野達也室長らと共同)が進められてきた。そして今回、結晶系の高精度なモデリング手法が新たに開発され、ナノ-バイオ境界系へのアプローチが可能となったというわけである。
FMO法以外にも、大規模系の定番手法としては「古典分子動力学(MD)法」もしくは「古典/量子論混成(QM/MM)モデリング」もあるが、それぞれ弱点があった。古典MD法では電荷移動や化学反応などの精密な現象を扱うことができないし、QM/MMにおいては今回の系のようにペプチドと固体側の広範囲にわたる界面を量子論(QM)で扱わなければならない場合、実際の計算は極めて難しくなってしまう。また、固体物理で広く利用されている「密度汎関数計算」(CPMDなど)も、系の非周期性や分散力の欠如の点から適用は現時点では困難である。
具体的に研究グループが今回成功したのは、FMO4法をシリカ(酸化ケイ素)表面への6残基のアミノ酸からなるペプチドの結合状態のモデリングに応用することだ。Si-O結合の3次元的ネットワークを持つ固体シリカの扱いは、FMO4法による高精度化とシリカ結晶の3次元的な分割手法の開発で可能になったものである。なお、3次元的に配置したSi-O結合を持つシリカ結晶の切断では、結晶のユニットセルを基本構造としたフラグメントに分割する。形式電荷やフラグメントサイズが均一になるように配置される形だ。
対象とした6残基ペプチドは、芝清隆氏の研究グループによって開発された、チタン、銀、およびケイ素の酸化物表面に対してのみ特異的に結合する12残基のペプチドの前半部分、表面固着能を決しているとされる6残基「Arg1-Lys2-Leu3-Pro4-Asp5-Ala6(RKLPDA)」だ(画像4)。そして、シリコン原子を257個含むシリカのクラスターモデル(画像5)にこのRKLPDA片を組み合わせた複合系(水和も考慮)に対し、分散力を取り込める2次の摂動論(MP2)レベルで大規模な計算が行われた。

|

|
|
画像4。RKLPDAのモデル(プレス発表用資料から抜粋) |
画像5。RKLPDAとシリカのクラスターモデル(プレス発表用資料から抜粋) |
実行にはノード当たり12個コア、それを168ノード持つFOCUSスパコンを用い、特に時間のかかる4体フラグメントにおける処理時間を短縮するために「コレスキー分解(CDAM-MP2法)」が使用されている(FMO-MP2/6-31Gレベル)。系の4乗に比例する4中心の2電子積分をコレスキー分解によって近似的に3中心で評価する技法だ。高速でありながら化学的な精度は担保されるのが特徴で、FMO4のMP2計算では、コストは半分程度に抑えられるのが特徴。
また相互作用エネルギー解析では、フラグメント間の遮蔽効果を統計的に考慮した新しい「SCIFIE(Statistically Corrected Inter-Fragment Interaction Energy)法」も採用された。SCIFIEとは、古典統計力学の多体論的な手法を用いてIFIEからフラグメント間の実効ポテンシャルを求める方法である。イオン性が強い系ではIFIEが過大評価される傾向にあるが、SCIFIEでは統計的な補正手法により遠方のフラグメントで期待される減衰効果を含めた値を得ることが可能だ。
なお、画像6と7はIFIEによる相互作用を可視化したものだが、シリカ表面への結合ではRKLPDAの中で荷電したArg1、Lys2、Asp5の3つのアミノ酸残基がとりわけ重要であることが確認された。こうした情報は、ペプチドの改変や最適化に対して計算の立場から有益な指針を提供するものといえるという。一方、シリカ側もペプチドの結合によって表面だけでなく奥も分極されており、固体側をより簡便な小型のクラスターモデルで近似することができないことも判明した。

|

|
|
水和したシリカ-ペプチド複合系の相互作用の可視化。画像6(左)はペプチド側、画像7はシリカ側に注目して合算している。赤は結合による安定化、青は不安定化を示す(プレス発表用資料から抜粋) |
|
今後、研究チームは、今回成功したFMO4計算によるペプチドとシリカ表面のモデル計算を発展させることを計画中だ。別種のペプチドを組み合わせて相互作用のパターンの差異を見ることに対し、「興味深いこと」だという。これは、表面に強く結合するアミノ酸残基の特定に役立つのはもちろん、むしろ吸着しない・させないペプチドを設計するのにも使うことが可能だ。
また、固体結晶の欠損やドープなどのモデリングやシリカ以外の固体表面でアルミナやゼオライトなどを対象にすることも考えているという。さらに、酸化物に留まらず各種のエンジニアリングポリマーやダイヤモンド薄膜などを扱うことも視野に入れているとした。計算資源としては、「京」や全国9つの大学が保有するベクトル型を含むスーパーコンピュータや大規模ストレージシステムをネットワークで結んだ「HPCI(High Performance Computing Infrastructure)」を用いる予定としている。
さらに、今回の計算手法は、FMO法の地球科学分野への応用に道を開くものでもあるという。シリカは、石英などの鉱物の基本構成要素であり、水和条件下における鉱物表面への各種イオンの吸着・脱着は重要な化学プロセスだが、電子状態計算による理解はこれまではほとんどなされていなかった。しかし、このFMO4計算の技術を発展させることにより、熱力学的なパラメータを含めた詳細な描像が得られるようになると期待されるとする(画像8・9)。

|

|
|
シリカ表面とイオンの相互作用の可視化。画像8(左)ナトリウムイオン。画像9は塩素イオン。赤は結合による安定化、青は不安定化を示す(プレス発表用資料から抜粋) |
|
今回の成果に対して研究チームは、これまで主に生化学・生物物理化学・薬学分野に限られてきたFMO法の適用範囲を拡大することから、ブレークスルーと位置づけられるとした。