京都大学は12月10日、滋賀医科大学の協力を得て、タンパク質分解異常に着目した遺伝子改変マウスの作製により、原因が未だに多くの謎に包まれ、治療法が確立されていない神経難病の1つである「筋萎縮性側索硬化症(ALS)」の疾患再現に成功したと発表した。
成果は、京大 医学研究科高橋良輔教授、同・田代善崇教務補佐員、滋賀医科大 分子神経科学研究センターの漆谷真准教授らの共同研究グループによるもの。研究の詳細な内容は、米国科学誌「The Journal of Biological Chemistry」に掲載された。
ALSは進行性の筋肉の萎縮と筋力低下を主症状とし、3年から5年程度で呼吸不全によって死に至る最難治性神経変性疾患の1種であり、現在までのところ有効な治療法は開発されていない。
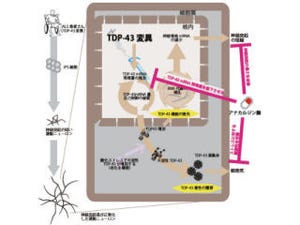
近年、パーキンソン病など多くの神経難病では病巣に異常タンパク質が蓄積することが知られており、ALSにおいても、「TDP-43」や「Fused in Sarcoma(FUS)」、「optineurin」などの蓄積が徐々に明らかになってきた。
異常タンパク質は細胞内のタンパク質分解機構である「ユビキチン・プロテアソーム」系や「オートファジー(自食作用)・リソソーム」系で分解されるため、これらの機能障害が病因仮説として挙げられていたが、ALSでは如何なる分解障害によって病的なタンパク質が蓄積するのかについて一定の見解がなく、動物で証明した研究も過去に存在しなかったのである。
そこで研究グループは今回、ALSの主要な病巣である「脊髄運動ニューロン」に特異的にプロテアソームとオートファジーに必須な分子を欠損するマウスをそれぞれ作製し、各々のタンパク質分解機構の障害とALS症状や病理学的異常の有無を詳細に調べたというわけだ。
プロテアソーム障害マウスは、8週齢以降に振戦(ふるえ)様症状や尻尾吊り下げ時の下肢伸展反射の低下(画像1・2)に始まる下肢の麻痺を呈し始め、徐々に歩行不能となるというALSと類似の症候を示した。

|

|
|
画像1。下肢進展反射の低下。左は正常なマウスで、右がプロテアソーム障害マウス |
画像2。秋冷別の運動ニューロン数。プロテアソーム障害マウスは週齢が上がると目に見えて減っていく |
病理学解析では、プロテアソーム障害マウスの運動ニューロン数が進行性に減少し(画像3)、「ミクログリア」や「アストログリア」の増殖とALSに特徴的な「chromatolytic neuron」や「basophilic inclusion」を認めた。さらに、家族性ALSで遺伝子突然変異が知られているTDP-43、FUS、optineurin、「ubiquilin2」タンパク質が著明に蓄積していたのである(画像4)。
これに対して、オートファジー障害マウスの運動ニューロンではいくつかの特徴は見られたものの孤発性ALSとは異なる変化であり、ニューロン数の変化はなく、マウスの寿命に近い2年齢まで運動機能は正常だった。

|

|
|
画像3。運動ニューロン数の低下 |
画像4。孤発性ALSと類似の病理所見 |
タンパク質分解の2大機構の内、運動ニューロンにおけるプロテアソームの障害が孤発性ALSの発症に関わることが、今回の研究で直接的に証明された形だ。蓄積タンパク質の解析により、ALSの病態機序の解明と治療法の確立が期待できるという。さらに、今回作製されたマウスは孤発性ALSの新たなモデル動物として、治療開発研究を促進することが期待できるとした。
研究グループは今後、今回の遺伝子改変マウスを用いて、病態機序の解明や、治療効果が得られる薬の検索などを行い、ALSの根本治療に向けて研究を行っていくとしている。